Note
Go to the end to download the full example code.
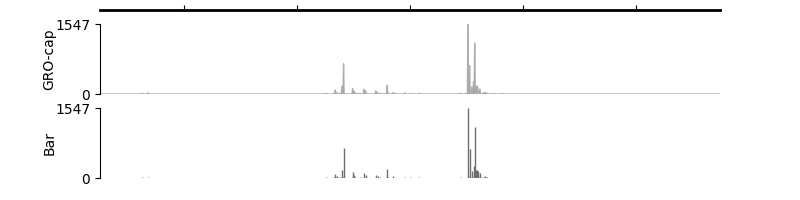
Paired Strandless BigWig Track#
Create a PairedStrandlessTracks to show signals
on the forward strand (pl_track) and the reverse strand (mn_track) in a strandless-manner.
This can be useful for presenting data from TSS-assays like GRO-cap/PRO-cap, NETCAGE, etc
and NascentTranscript-assays like PRO-seq, TT-seq, etc.
Note: The final signal values will be basepair-wise sum of the values on the forward strand and the absolute values on the reverse strand.
array([<Axes: title={'center': 'chr1'}, ylabel='GRO-cap'>,
<Axes: ylabel='Bar'>], dtype=object)
from pygv.viewer import GenomeViewer
from pygv.tracks.bigwig_track import PairedStrandlessTrack
gv = GenomeViewer()
# draw positive and negative values independently
independent_track = PairedStrandlessTrack(
"../examples/data/K562_GROcap_hg38_pl.chr1.bw",
"../examples/data/K562_GROcap_hg38_mn.chr1.bw",
name="GRO-cap", y_label_rotation="vertical", y_label_ha="center")
gv.add_track(independent_track)
# Bar plot
bar_track = PairedStrandlessTrack(
"../examples/data/K562_GROcap_hg38_pl.chr1.bw",
"../examples/data/K562_GROcap_hg38_mn.chr1.bw",
plot_type="bar", name="Bar", y_label_rotation="vertical", y_label_ha="center")
gv.add_track(bar_track)
gv.plot("chr1", 201954851, 201955948)
Total running time of the script: (0 minutes 0.124 seconds)